
细胞转染是指利用各种物理、化学、生物学方法,将外源分子(DNA、RNA 、mRNA等)导入真核细胞内,来改变细胞的特性,从而达到改造细胞的目的,它是实现细胞基因功能和蛋白质表达研究的重要工具。
根据外源核酸是否整合到宿主染色体上,细胞转染可以分为瞬转和稳转。根据转染方式又可以分为:物理转染法、化学转染法及生物转染法,这三大类又包含了许多不同的转染技术和操作方法。
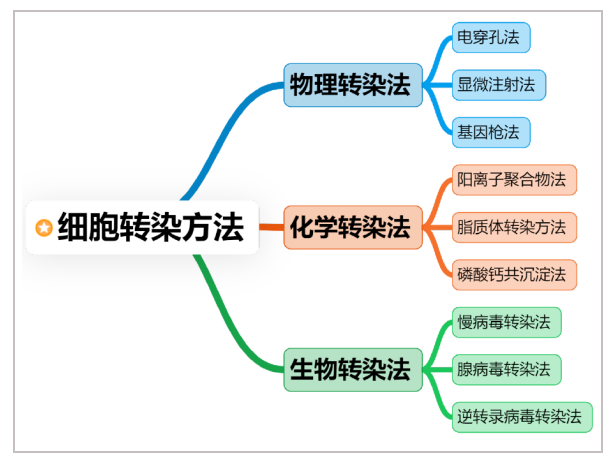
图1:细胞转染方法
别慌!小优在这里为大家整理了一份转染秘籍,以目前运用最为广泛的三大转染技术——电穿孔法、阳离子聚合物转染法、慢病毒转染法为例,详细介绍了它们的原理、操作方法和提高转染效率的优化方案,以及如何选择合适的转染方案,帮助大家快速提升细胞转染效率,强烈建议小伙伴们收藏并转发~
PART 1 电穿孔法
01 电转法原理
电穿孔法,又叫电转法,利用电脉冲在细胞膜上形成暂时的孔,使核酸物质能穿过孔进入细胞。首先,使用带电荷的转染试剂将目的基因或者质粒包裹起来,形成转染复合物;然后,细胞在仪器的高场强电脉冲处理之后,会在一侧形成电压差异,从而使得目的基因以类似电泳的方式进入到细胞内。
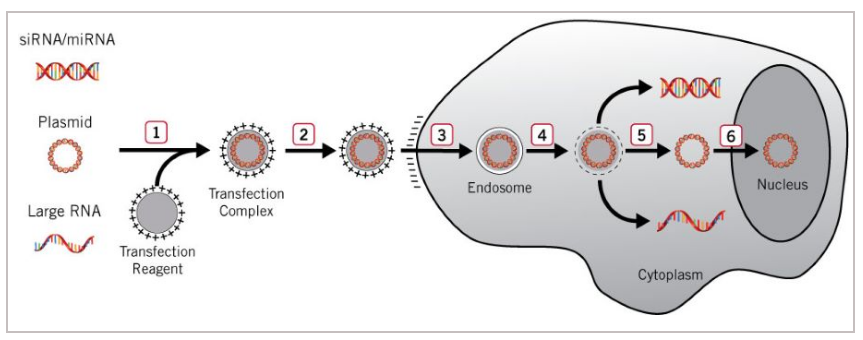
图2:电转法原理图
02 Nucleofector™ 技术操作方法
电转法操作的重点在于控制电压和通电时间,如果电压过强,或者脉冲时间太久,都可能会导致细胞死亡;但是,如果减少电压和脉冲时间,又会降低转染效率,尤其是在对原代细胞、干细胞、免疫细胞等难转染的细胞进行转染时,想要同时兼顾转染效率和细胞存活率是非常困难的。
为了克服传统电转法的这些弊端,近年来科学家们开发出来的电转技术层出不穷,备受好评的便是Nucleofector™ 技术。Nucleofector™技术是Lonza公司的专利创新技术,通过电击在细胞膜上开个小孔,综合各种特定细胞转染程序与转染液的作用,核酸底物不仅可以进入细胞质,还可通过核膜进入细胞核。相较于传统电转技术,Nucleofector™技术的细胞转染率最高可达99%,且实现转染不依赖于细胞的分裂,是全球公认的高效转染技术。
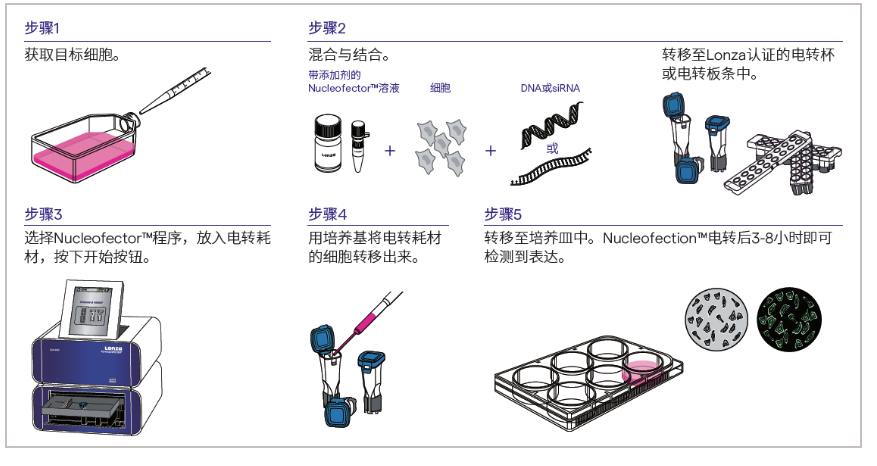
以转染DNA为例,Nucleofector™技术的操作步骤:
1、收获目的细胞
贴壁细胞用胰酶消化收获细胞。按照转染需要的细胞量进行计数,如20 µL电转体系可转1x104-1x106个细胞,100 µL电转体系可转1x105-1x107个细胞;
2、加入质粒DNA并混匀
低速离心,尽量吸去细胞上清,用电转Buffer重悬细胞,加入转染杯,加入准备好的质粒DNA,轻轻混匀,避免产生气泡;
3、启动电转程序
将转染杯放入转染仪,按键选择相应程序,按"start"键开始转染,等待指示图标显示绿色“+”号,即可完成实验;
4、细胞培养
吸去电转Buffer,加入预热好的培养基,将细胞转移至培养皿中继续培养。若转染的质粒为试剂盒内的阳性对照质粒,一般在实验结束后7-8 h内可在荧光显微镜下观察到实验结果。
PART 2 阳离子聚合物转染法
01 阳离子聚合物转染法原理
带正电的聚合物与核酸带负电的磷酸基团形成带正电的复合物后,与细胞表面带负电的蛋白多糖相互作用,并通过内吞作用进入细胞。阳离子聚合物和阳离子脂质体最大的区别在于阳离子聚合物不含疏水部分而完全溶于水,可以方便地进行化学修饰,且不会像脂质体一样破坏细胞结构,所以毒性相对较低。
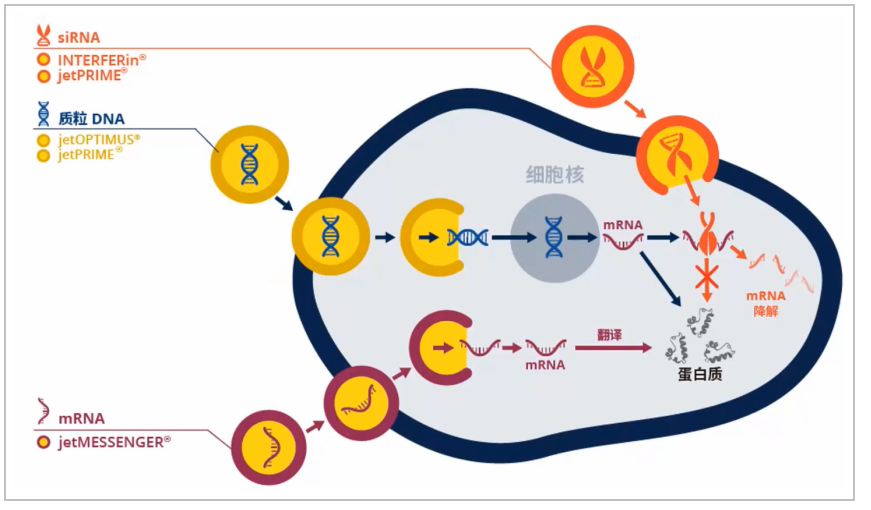
图4:阳离子聚合物转染法原理图
02 阳离子聚合物转染法操作方法
以jetPRIME®进行DNA转染为例:
1、为了达到最佳的DNA转染条件,建议在细胞汇合度为60%-80%时进行转染。通常,用6孔板进行实验时,转染前24h在每个2mL的孔里接种大约2x105个细胞;
2、将2µg DNA稀释到200µL jetPRIME®缓冲液中,通过涡旋混合;
3、涡旋震荡jetPRIME®试剂5秒,使用前短暂离心;
4、在DNA中添加4µL jetPRIME®试剂,涡旋震荡1秒钟,短暂离心;
5、在室温下孵育10分钟;
6、将200µL转染混合物逐滴加到含血清培养基的细胞上,确保均匀分布滴加;
7、轻轻地水平摇晃培养板,置于在37°C孵育;
8、如果需要,在转染后4小时至24小时用正常培养基替换转染培养基,并根据需要进行分析;
9、24小时后进行检测。
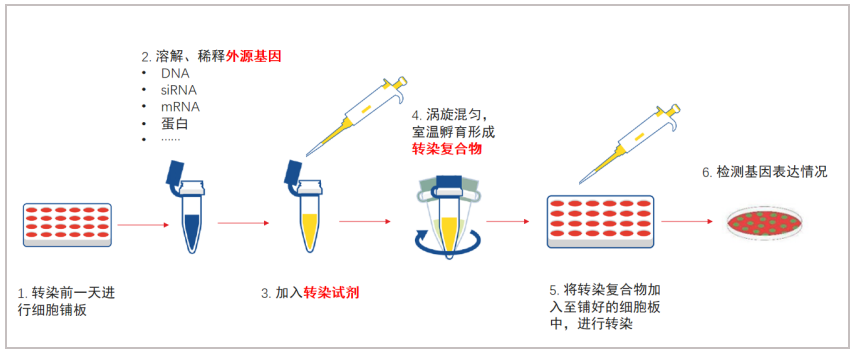
图5:阳离子聚合物转染法的操作步骤
PART 3 慢病毒转染法
01 慢病毒转染法原理
常见的载体包括慢病毒、腺病毒和逆转录病毒,转染效率特别高,尤其是难以转染的原代细胞、活体细胞,同时具有细胞毒性很低的优势。其中,慢病毒因其高稳定性,被广泛用于哺乳动物细胞体内外的转染实验中。
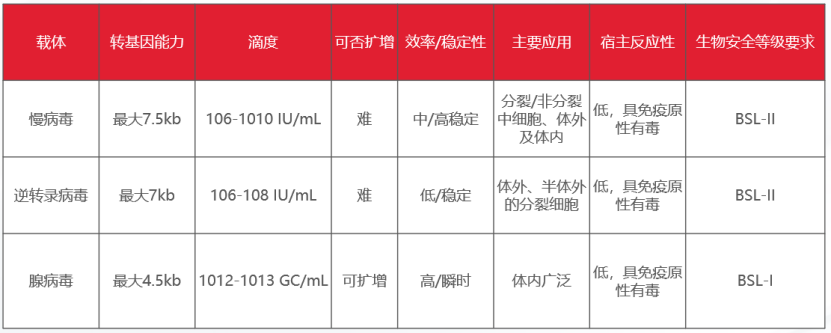
表1:三种病毒载体的对比
02 慢病毒转染法操作方法
以PEIpro®转染DNA为例:
1、 在细胞接种时,需要调整细胞密度,以便在转染时,细胞达到指数生长阶段,并且活细胞密度(VCD)约为2-2.5x106 cells/mL;
2、 转染当天,测量细胞密度并确定转染参数(每百万细胞的 DNA 用量和PEIpro®体积)。例如,我们假设 VCD 为每毫升细胞培养基中 2x106 cells/mL;
3、 在无血清培养基(无添加剂)中稀释 2mg DNA 至最终体积为 50mL,轻轻涡旋混匀;
4、 迅速涡旋 PEIpro®试剂;
5、 在无血清培养基(无添加剂)中稀释 2mL PEIpro®至最终体积 50mL,轻轻涡旋混匀;
6、 立即将 50mL PEIpro®溶液添加到 50mL DNA 溶液中;
7、 立即混合溶液,建议短暂地涡旋溶液或将离心管轻柔颠倒 3~4 次混匀;
8、 在静止和室温下孵育复合物 15 分钟;
9、 向细胞中加入 100 mL PEIpro®/DNA 混合物;
10、在适当的温度、转速和CO2水平(例如 37℃、130rpm、8%)下培养细胞,并收获病毒。

图6:慢病毒转染悬浮细胞的操作步骤
PART 4 影响细胞转染的因素以及优化方案
01 细胞状态
在进行转染时,我们需要选择合适状态的细胞,一般原代细胞可以传1-2次后使用;尽量挑选迭代次数少的细胞,如果是冻存细胞,建议复苏1-2h后使用。细胞污染对细胞状态的影响很大,受到细菌、真菌或支原体污染的细胞都会导致转染效率降低、细胞死亡率增加。
细菌污染
细菌是一种原核细胞微生物,其大小以微米(μm)计,细菌污染会导致细胞内颗粒增多、增粗,最后变圆脱落死亡,严重影响了细胞的转染效率。抗生素对杀灭细菌较有效,联合用药比单独用药效果好。在污染早期,预防用药一般用双抗生素,污染后清除用药需采用大于常用量5-10倍的冲洗法,加药后作用12-24小时,再换常规培养液。
真菌污染
发生真菌污染时,可以观察到在培养培养液表面形成白色或黄色漂浮物,在高倍镜下可见明显的丝状、管状或是树枝状的菌丝纵横交错在细胞间。一旦发生真菌污染,那么细胞转染实验就宣告失败了,必须直接将污染物移除培养间丢弃,切不可在培养间内打开培养器皿,因此,做好预防尤其重要。
支原体污染
支原体污染,在显微镜下很难观察到,同时,它不受抗生素影响。支原体污染尤其会影响细胞脂质体转染和细胞电转染效率,因此支原体污染的检测和处理非常重要。如果污染细胞价值不大,弃之;有细胞株留存的或可购置的,可在寻找原因后彻底消毒操作室,复苏或重新购置细胞,再进行培养。若污染的细胞价值较大,难于重新得到,可采取如下办法清除:
1、加温处理:将污染的组织培养物放在41℃培养18小时,可杀死支原体,但对细胞有不良影响;
2、清洗纯化法:其原理是利用离心力、细胞、微生物质量和悬液的浮力差达到清除支原体的目的;
3、药物处理:敏感抗生素;
4、支原体清除试剂盒是解决细胞培养中支原体污染严重问题的有效方法。
02 内毒素
内毒素是革兰氏阴性细菌细胞壁中的脂多糖,当细菌死亡溶解或用人工破坏细胞后,内毒素会释放出来。如果细胞的内毒素水平过高,那么在进行转染时,会导致细胞死亡率增加,大大降低转染效率。因此,提前和定期给我们的细胞做内毒素检测,是转染前一项非常重要的工作。
03 培养基
转染培养基中,如果含有抗生素会影响细胞转染效率,因为转染试剂本身对细胞就有损伤,这时培养基中的抗生素会进一步损伤细胞,因此我们所有转染过程都建议使用不含抗生素的培养基或PBS。此外,某些细胞类型(如神经细胞、多能性干细胞等)对血清敏感,需要用无血清培养体系。
04 细胞铺板密度
不同的转染试剂,对细胞铺板密度的要求不尽相同。因此,在进行不同核酸或不同细胞系的转染时,需要根据说明书再次优化实验条件。一般来说,贴壁细胞密度为70%-90%,悬浮细胞密度为2×106-4×106细胞/ml时效果较好,确保转染时细胞没有长满或处于静止期。
05 试剂比例
通常转染不同的分子都有不同的最佳比例,如果DNA转染试剂与DNA质粒复合物配比不合适,就会影响转染效率。因此我们在实验开始之前,需要仔细阅读说明书或者参考文献,使用最佳配比浓度,或者进行预实验。
06 细胞换液
使用传统转染试剂,转染后4-6h后必须进行换液,否则会由于转染试剂毒性而引起细胞死亡。


